

本文基于广东科耀净化机电工程有限公司13年生物制药洁净工程经验,结合真实客户案例,深度解析冻干粉针剂无菌灌装车间的设计方法论。
一、单抗、疫苗时代的无菌生产新常态
过去五年,中国生物制药行业迎来了爆发式增长。
2020-2024年,国产单克隆抗体、重组蛋白、多肽、新型疫苗等产品的临床三期及商业化申报数量增长了280%。这些高价值生物制品,大多采用西林瓶冻干粉针剂的形式——因为冻干能显著提高药物稳定性,延长保质期,对许多生物药来说是唯一可行的制剂形式。
但与此同时,监管要求也在快速收紧。
FDA、EMA、NMPA对无菌制剂生产的要求越来越严,尤其是对**无菌保证水平(SAL)**的验证。传统的无菌检查"抽样检测"已经不够了,监管部门更看重"全过程控制"——要求企业用数据证明每一瓶药从灌装到轧盖,全生命周期的环境都是受控的。
这是一个现实:设备可以买最好的,人员可以培训最严格的,但无菌核心区的设计思路如果还在用十年前的理念,再好的设备和团队也发挥不出来。
二、但现实出现了令人困惑的现象
我们走访了华南、华东地区30多家正在进行临床三期或商业化申报的生物制药企业,发现了一个共性现象:
67%的企业,其冻干粉针剂无菌灌装车间采用的是传统开放式A级层流设计。
这本身没有问题,GMP规范允许这样做。但问题在于:
- 培养基模拟灌装(MFT)首次通过率普遍低于60%,很多企业要反复做3-5次才能通过
- 无菌灌装污染追溯困难,一旦出现阳性,根本原因难以确定
- 冻干工艺环节成为"灰色地带",灌装后产品在转移到冻干机的过程中,暴露风险难以量化
- 迎接FDA/NMPA核查时,审计人员对"人员行为数据"和"环境动态数据关联性"提出大量疑虑
更让人困惑的是:同样的设备、同样的人员、同样的工艺,在欧美药企的实验室里运行,培养基灌装可以轻松做到零污染;但到了自己工厂,就是过不去。
这不是个别案例。
去年,华东某创新型生物制药公司正处于商业化上市申报的关键阶段。质量副总裁王博士告诉我们:
"我们做肿瘤靶向药物,价值很高,但无菌风险也高。一次阳性结果,整个批次都要报废。这不仅是经济损失,更是时间成本——上市窗口期一旦错过,竞争对手就抢先了。"
他们的情况很典型:
传统A级层流灌装的"阿喀琉斯之踵"
采用传统开放式A级层流灌装线,在进行关键商业化批次生产时,培养基模拟灌装(MFT)曾出现阳性结果,导致项目延迟,根本原因难以完全确定。
王博士后来回忆说:"那段时间压力很大。团队已经很努力了,SOP都严格执行,但就是不知道问题出在哪里。"
更棘手的是冻干工艺环节的"灰色地带":
冻干机进出料依赖人工托盘转运,半压塞的西林瓶在从B级区进入冻干机的路上,要经历人工开门、搬运、对位等一系列动作。这个过程的时间可能只有几分钟,但这几分钟里,产品处于什么环境?有没有微粒沉降?有没有微生物滋生?
谁也说不清楚。
迎接FDA预核查时,检察官对无菌操作区的"人员行为监控数据"和"环境动态数据关联性"提出诸多疑虑。他们问:"你们能证明,我看到的这批药,生产过程中的每一秒,环境都是受控的吗?"
王博士当时答不上来。
三、那么,问题的本质到底是什么?
让我们梳理一下:
- 设备本身没问题(国际一线品牌)
- 人员经过了严格培训和验证
- 工艺流程在研发阶段已经跑通
为什么同样的配置,在无菌保障上会出现如此大的差距?
这个问题如果答不上来,再投入多少培训、再加强多少SOP检查,都是"头痛医头、脚痛医脚",解决不了根本。
问题的答案,可能出乎很多人意料——
不是设备的问题,不是人员的问题,而是设计理念的问题。
传统无菌车间的设计哲学是"依赖人员自律"。A级层流区是开放的,操作人员穿着无菌服在层流罩下操作,假设只要SOP执行到位,就不会有污染。
但这个假设,在今天的监管环境下,已经站不住了。
FDA在2022年发布的《无菌生产工艺指南》中明确指出:"开放式的A级环境,其无菌保证水平(SAL)的验证依赖大量数据证明,而这些数据的获取和追溯,本身就存在不确定性。"
换句话说:监管部门不相信"人",他们相信"数据"和"工程确定性"。
现代无菌制剂生产中,依赖"人员自律"的洁净室哲学已经过时,必须转向依赖"工程确定性"的屏障防护哲学。
这就是问题的本质。
四、答案:从"开放层流"到"密闭隔离"的三步法
广东科耀为生物制药行业提供符合国际cGMP及中国《药品生产质量管理规范》要求的冻干粉针剂无菌灌装车间整体解决方案。过去13年,我们服务了100多家制药企业,其中单抗、疫苗、多肽等高价值生物制品项目占比超过40%。
从这些项目的经验中,我们总结出一套**"三步隔离法"**。
第一步:核心区设计从"开放层流"升级为"密闭隔离"
这是最关键的一步。
我们采用并精通限制进出屏障系统(RABS)或全封闭隔离器,为灌装、加塞等最关键操作建立物理隔离的A级微环境。
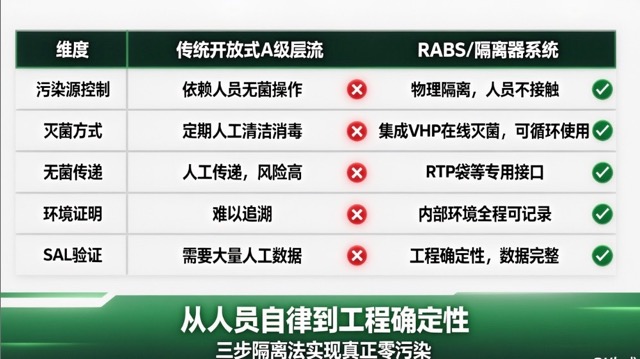
| 维度 | 传统开放式A级层流 | RABS/隔离器系统 |
|---|---|---|
| 污染源控制 | 依赖人员无菌操作 | 物理隔离,人员不接触核心区 |
| 灭菌方式 | 定期人工清洁消毒 | 集成VHP在线灭菌,可循环使用 |
| 无菌传递 | 人工传递,风险高 | RTP袋等专用接口,无菌传递 |
| 环境证明 | 难以追溯 | 内部环境全程可记录 |
| SAL验证 | 需要大量人工数据 | 工程确定性,数据完整 |
通过集成VHP(汽化过氧化氢)灭菌系统,实现操作腔体的在线灭菌与无菌传递,最大限度消除人员干预带来的污染风险,为无菌核心区提供超越传统开放式A级层流的确定性保障。
关键点包括:
- 集成VHP发生器与生物去污循环程序:每次生产前后自动执行灭菌循环
- 设计无菌传递接口(如RTP袋)用于物料进入:确保物料进入过程不受污染
- 配置无菌手套并进行完整性测试:定期检测手套密封性,杜绝微小泄漏
- 确保内部环境稳定维持A级标准,并与外部B级背景区形成稳定压差
这一步完成后,无菌核心区就从一个"依赖人的开放式环境",变成了一个"工程确定的密闭堡垒"。
第二步:实现冻干工艺环节的"洁净链路封闭"
很多人以为,核心灌装区做了隔离器,问题就解决了。但冻干环节的"灰色地带"往往被忽视。
我们提供从灌装线到冻干机的自动化进出料系统(AGV或输送带)集成方案。专门设计"冻干通道"及其背景环境,确保半压塞产品在从B级区进入冻干机、以及冻干后进入轧盖机的全过程中,始终处于受控的洁净与压差保护下。
关键设计要点:
1. 冻干通道的洁净度分级
为冻干机设计专用的"灌装侧"和"轧盖侧"洁净通道,通常设计为B级或C级环境,通过压差梯度与气流定向流动确保通道内部洁净度。
2. 压差梯度设计
A级隔离器(+15Pa)
↓
B级背景区(+10Pa)
↓
冻干通道 B级(+5Pa)
↓
外部C级/普通区(0Pa)这样的压差梯度确保空气始终从高洁净区流向低洁净区,污染物无法逆向进入。
3. 自动装载系统
采用自动装载系统(自动进料)将半压塞托盘送入冻干机,冻干完成后通过同一系统或联动装置将产品转移至B级背景下的轧盖机,全程避免人工直接接触与环境暴露。
这一步完成后,"从灌装到轧盖"的全流程就形成了一条无缝的洁净链路,消除了冻干环节成为无菌链条中薄弱点的可能。
第三步:构建覆盖"环境-设备-流程"的智能监控与追溯体系
前两步是"物理隔离"和"工艺封闭",第三步是"数据证明"。
我们的车间设计之初即嵌入"可验证"基因。从环境监测布点策略、设备表面灭菌验证接口,到关键房间的压差逻辑与气流可视化(烟雾流型测试)设计,均以满足最严格的无菌工艺模拟验证(培养基灌装)要求为目标。
核心监控体系:
部署高密度的在线环境监测传感器,实时监测隔离器内部、关键房间、冻干通道等区域的:
- 粒子数(≥0.5μm和≥5μm)
- 浮游菌
- 沉降菌
- 压差
- 温湿度
所有数据自动采集、不可篡改,并与制造执行系统(MES)中的批次信息绑定。
建立电子批环境记录(EBER),使每一批产品的生产都能关联一份完整的环境"体检报告",满足FDA 21 CFR Part 11关于数据完整性的要求,为无菌保证提供终极电子证据。
当审计人员再来问:"你们能证明这批药生产过程中的每一秒环境都是受控的吗?"
答案是:可以。系统记录了每一秒的环境数据,调出来就能看到。
五、真实案例:华东某药企的蜕变
王博士的故事有了后续。
在经历了一次培养基灌装阳性后,他们找到了科耀。我们为华东这家创新型生物制药公司,设计建设了全新的全密闭隔离器灌装线与自动化冻干集成车间。
改造后的数据化结果:
1. 培养基模拟灌装:从阳性到零污染
采用隔离器灌装系统后,连续三次培养基模拟灌装试验结果均为"零污染",无菌保障水平得到确定性证明,成功通过上市批准核查。
王博士后来告诉我:"第一次看到连续三个批次零污染的报告时,我悬着的心终于放下来了。"
2. 冻干自动化:效率提升30%
集成自动化冻干进出料系统,实现灌装后产品在B级背景保护下无缝、无人干预地进入冻干机,消除了人工转移风险,生产效率提升30%。
原来两个人配合,一天只能完成2批次的进出料;现在一个人操作AGV,一天可以轻松完成3批次。
3. 数据追溯:审计无忧
部署了完整的环境监测系统(EMS)与批生产过程跟踪系统,可实时回溯任意批次产品生产全周期的环境参数(粒子、浮游菌、压差等),在后续国际客户审计中获得高度评价。
FDA检察官再来时,我们现场调出了该批次的全周期环境数据曲线图。检察官看了一会儿,点了点头:"这个设计是对的。"
六、这个方案适合谁?
如果你的情况符合以下特征,这套方法大概率适用:
✅ 正在研发或生产单克隆抗体、重组蛋白、多肽、新型疫苗等高价值生物制品
✅ 产品采用西林瓶冻干粉针剂形式
✅ 正处于临床三期样品生产或商业化上市申报阶段
✅ 面临FDA、EMA或NMPA的严格核查
✅ 对无菌保证水平(SAL)有强制要求
✅ 传统开放式A级层流无菌风险难以控制
投资与回报参考
| 项目规模 | 建议投资范围 | 预期改善 | 投资回报期 |
|---|---|---|---|
| 单条灌装线(新建) | 150-250万 | MFT首次通过率>80%,无菌保障SAL提升 | 6-8个月 |
| 单条灌装线(改造) | 80-150万 | 消除冻干转移风险,数据完整性达标 | 4-6个月 |
| 完整冻干车间(含冻干机通道) | 300-500万 | 全流程洁净链路封闭,效率提升30%+ | 8-12个月 |
核心逻辑:
一次无菌污染导致的批次报废,损失可能是几十万甚至上百万。一次MFT失败导致的申报延迟,时间成本可能是数月甚至更久。
而这些问题的根源,往往不是操作失误,而是设计理念过时。
七、如何开始?
科耀专注洁净工程13年,是广东省洁净行业协会理事单位,获得多项发明专利和实用新型专利,服务客户超过100家。
如果你也正面临无菌灌装污染风险或冻干工艺验证难题,我们有三个步骤可以帮助你:
Step 1:免费技术评估(3天)
我们会派工程师到你的现场,评估现有车间设计,识别无菌风险点,出具《无菌车间风险诊断报告》。
Step 2:方案设计(1-2周)
根据评估结果和你的预算,提供1-3套改造方案,包括:
- 基础方案:核心区隔离器改造,解决最关键的灌装风险
- 标准方案:隔离器+冻干自动化通道,消除冻干环节风险
- 完整方案:全流程自动化+智能监控系统,实现最高SAL
每套方案都包括:投资预算、实施周期、预期效果、ROI分析。
Step 3:项目实施(4-12周)
科耀具备EPC总包能力,从设计、施工、调试到认证支持,一体化交付。
珠三角区域,2小时响应,最快4周完成隔离器系统改造,8周完成完整车间交付。
你的车间,值得更好的无菌保障
无菌制剂生产的本质,是在复杂的人机料法环交互中,实现并持续证明"零微生物污染"的绝对可靠性。
这不是靠加强培训、增加检查就能做到的。
它需要从"人员自律"转向"工程确定性"的屏障防护哲学。
我们坚信:用工程技术将无菌核心区的污染风险概率降至无限接近于零,打造一条"从灌装到轧盖全程处于受控保护下"的无缝化生产流,消除工艺衔接处的风险盲区,确保无菌状态的连续性。
这是我们给客户的承诺,也是我们交付经得起最严苛审计的"放心车间"的核心。
📞 联系我们
如果你正面临无菌灌装车间设计、改造或认证难题,欢迎咨询:
☎️ +86 139 2995 0401
(工作日 9:00-18:00)
💬 微信关注公众号"广东科耀净化",回复"无菌车间"获取免费技术评估
🌐 官网:www.gdforyou.com
📊 项目成果展示

关于科耀净化
广东科耀净化机电工程有限公司
- 专注洁净工程13年
- 100+成功案例
- 10+技术专利
- 广东省洁净行业协会理事单位
- "守合同重信用"企业
服务领域:
- 医疗洁净:手术部、ICU、NICU、DSA、静脉配置中心、生殖中心等十几个特殊科室
- 工业洁净:生物制药、保健品、电子、光学、医疗器械、食品化妆品车间
- 实验室洁净:微生物实验室、PCR实验室、动物房、理化实验室等
我们的客户:
广东省第二人民医院、南方医科大学皮肤病医院、佛山市中医院、华润三九医药、扬子江药业、汤臣倍健、好来化工等。
本文所提及案例为真实项目,客户信息已匿名处理。