

Based on 13 years of biopharmaceutical cleanroom engineering experience from Guangdong Keyao Purification Electromechanical Engineering Co., Ltd., combined with real customer cases, this article provides an in-depth analysis of the design methodology for lyophilized powder injection sterile filling rooms.
I. The New Normal of Sterile Production in the Era of Monoclonal Antibodies and Vaccines
Over the past five years, China's biopharmaceutical industry has experienced explosive growth.
From 2020 to 2024, the number of Phase III clinical and commercial filing applications for domestic monoclonal antibodies, recombinant proteins, peptides, and new vaccines increased by 280%. These high-value biologics are mostly produced in the form of lyophilized powder injections in vials—because lyophilization can significantly improve drug stability and extend shelf life, making it the only viable dosage form for many biologics.
At the same time, regulatory requirements are rapidly tightening.
FDA, EMA, and NMPA are imposing increasingly strict requirements for sterile pharmaceutical production, especially for the verification of Sterility Assurance Level (SAL). Traditional "sampling-based" sterility testing is no longer sufficient. Regulators place greater emphasis on "full-process control"—requiring companies to use data to prove that every vial of medicine, from filling to capping, is produced in a controlled environment throughout its entire lifecycle.
This is a reality: you can buy the best equipment, train the most rigorous personnel, but if the design philosophy for the sterile core area still uses concepts from a decade ago, even the best equipment and teams won't be able to perform.
II. But Reality Presents a Confusing Phenomenon
We visited more than 30 biopharmaceutical companies in South and East China that are undergoing Phase III clinical or commercial filings and discovered a common phenomenon:
67% of companies' lyophilized powder injection sterile filling rooms use traditional open-style Grade A laminar flow design.
This itself is not a problem, as GMP regulations allow this approach. However, the issue lies in:
- Media Fill Test (MFT) first-time pass rates are generally below 60%, with many companies having to repeat tests 3-5 times before passing
- Sterile filling contamination traceability is difficult—once a positive result occurs, determining the root cause is extremely challenging
- The lyophilization process becomes a "gray zone"—exposure risks during the transfer of products to the lyophilizer after filling are difficult to quantify
- When facing FDA/NMPA inspections, auditors raise numerous concerns about the correlation between "personnel behavior data" and "dynamic environmental data"
What's more confusing is: with the same equipment, same personnel, and same process, in Western pharmaceutical company laboratories, media fills can easily achieve zero contamination; but in their own factories, it just doesn't work.
This is not an isolated case.
Last year, an innovative biopharmaceutical company in East China was in a critical phase of commercial上市 filing. Dr. Wang, Vice President of Quality, told us:
"We produce tumor-targeted drugs with high value, but the sterile risk is also high. One positive result means the entire batch must be scrapped. This is not just an economic loss, but also a time cost—once the上市 window is missed, competitors will take the lead."
Their situation is typical:
The "Achilles' Heel" of Traditional Grade A Laminar Flow Filling
Using a traditional open-style Grade A laminar flow filling line, during key commercial batch production, Media Fill Tests (MFT) showed positive results, causing project delays with root causes difficult to completely determine.
Dr. Wang later recalled: "The pressure was immense during that time. The team worked very hard and strictly followed SOPs, but we just couldn't figure out where the problem was."
Even more challenging is the "gray zone" in the lyophilization process:
Lyophilizer loading and unloading rely on manual tray transfer. Semi-stoppered vials experience a series of actions including manual door opening, carrying, and alignment when entering the lyophilizer from the Grade B area. This process may only take a few minutes, but during these few minutes, what environment is the product in? Are particles settling? Is microbial growth occurring?
No one can say for sure.
During the FDA pre-inspection, inspectors raised numerous concerns about the correlation between "personnel behavior monitoring data" and "dynamic environmental data" in the sterile operation area. They asked: "Can you prove that every second of the production process for this batch of medicine I see was in a controlled environment?"
Dr. Wang couldn't answer at the time.
III. So, What Is the Essence of the Problem?
Let's sort this out:
- The equipment itself is not the problem (top-tier international brands)
- Personnel have undergone rigorous training and verification
- The process workflow has been validated in the R&D phase
Why does the same configuration show such a huge gap in sterility assurance?
If this question cannot be answered, no matter how much more training or SOP inspections are strengthened, it's just "treating the symptoms, not the root cause."
The answer to this problem may surprise many people—
It's not an equipment problem, not a personnel problem, but a design philosophy problem.
The design philosophy of traditional sterile rooms is "relying on personnel self-discipline." Grade A laminar flow areas are open, with operators in sterile gowns working under laminar flow hoods, assuming that as long as SOPs are executed properly, there will be no contamination.
But this assumption no longer holds in today's regulatory environment.
The FDA's 2022 "Aseptic Processing Guide" explicitly states: "The verification of Sterility Assurance Level (SAL) in open Grade A environments relies on extensive data proof, and the acquisition and traceability of this data inherently have uncertainties."
In other words: regulators don't believe in "people"; they believe in "data" and "engineering determinism."
In modern sterile pharmaceutical production, the cleanroom philosophy of relying on "personnel self-discipline" is obsolete and must shift to a barrier protection philosophy of relying on "engineering determinism."
This is the essence of the problem.
IV. The Answer: Three-Step Method from "Open Laminar Flow" to "Closed Isolation"
Guangdong Keyao provides comprehensive solutions for lyophilized powder injection sterile filling rooms that comply with international cGMP and China's "Drug Manufacturing Quality Management Standards." Over the past 13 years, we have served more than 100 pharmaceutical companies, with monoclonal antibody, vaccine, and peptide high-value biologics projects accounting for over 40%.
From the experience of these projects, we have summarized a "Three-Step Isolation Method."
Step 1: Upgrade Core Area Design from "Open Laminar Flow" to "Closed Isolation"
This is the most critical step.
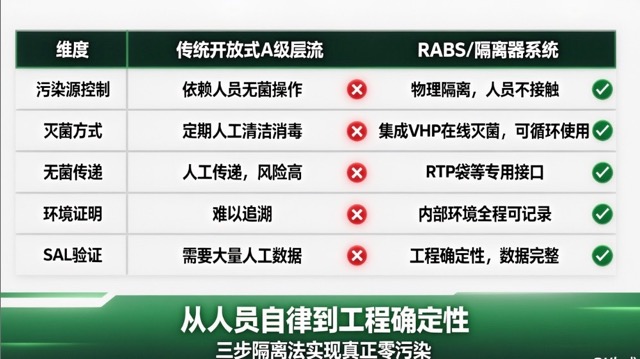
We specialize in using Restricted Access Barrier Systems (RABS) or fully enclosed isolators to establish physically isolated Grade A microenvironments for the most critical operations such as filling and stoppering.
| Dimension | Traditional Open Grade A Laminar Flow | RABS/Isolator System |
|---|---|---|
| Contamination Source Control | Relies on personnel aseptic operations | Physical isolation, personnel do not contact core area |
| Sterilization Method | Periodic manual cleaning and disinfection | Integrated VHP online sterilization, reusable |
| Aseptic Transfer | Manual transfer, high risk | RTP bags and other dedicated interfaces, aseptic transfer |
| Environmental Proof | Difficult to trace | Internal environment fully recordable |
| SAL Verification | Requires extensive manual data | Engineering determinism, complete data |
By integrating a VHP (vaporized hydrogen peroxide) sterilization system, we achieve online sterilization and aseptic transfer of the operation chamber, minimizing contamination risks from personnel intervention and providing deterministic sterility assurance for the sterile core area that surpasses traditional open Grade A laminar flow.
Key points include:
- Integrated VHP generator and biodecontamination cycle programs: Automatically execute sterilization cycles before and after each production
- Design aseptic transfer interfaces (such as RTP bags) for material entry: Ensure the material entry process is contamination-free
- Configure sterile gloves and perform integrity testing: Regularly test glove sealing to prevent micro-leaks
- Ensure internal environment stably maintains Grade A standards and forms stable pressure differential with the external Grade B background area
After this step is completed, the sterile core area transforms from a "personnel-dependent open environment" into an "engineering-deterministic closed fortress."
Step 2: Achieve "Clean Pathway Enclosure" for the Lyophilization Process
Many people assume that once the core filling area has an isolator, the problem is solved. However, the "gray zone" in the lyophilization process is often overlooked.
We provide integrated solutions for automated loading and unloading systems (AGV or conveyor belt) from the filling line to the lyophilizer. We specifically design "lyophilization channels" and their background environments to ensure that semi-stoppered products remain under controlled cleanliness and pressure protection during the entire process from entering the lyophilizer from the Grade B area to entering the capping machine after lyophilization.
Key design points:
1. Cleanliness Classification of Lyophilization Channels
Design dedicated "filling side" and "capping side" clean channels for the lyophilizer, typically designed as Grade B or C environments, ensuring internal cleanliness through pressure differential gradients and directional airflow.
2. Pressure Differential Gradient Design
Grade A Isolator (+15Pa)
↓
Grade B Background Area (+10Pa)
↓
Lyophilization Channel Grade B (+5Pa)
↓
External Grade C/Ordinary Area (0Pa)This pressure differential gradient ensures air always flows from high cleanliness areas to low cleanliness areas, preventing contaminants from entering in reverse.
3. Automatic Loading System
Adopt an automatic loading system to feed semi-stoppered trays into the lyophilizer. After lyophilization is complete, products are transferred via the same system or linked device to the capping machine under Grade B background, avoiding direct personnel contact and environmental exposure throughout.
After this step is completed, the "from filling to capping" full process forms a seamless clean pathway, eliminating the possibility of the lyophilization process becoming a weak link in the sterile chain.
Step 3: Build an Intelligent Monitoring and Traceability System Covering "Environment-Equipment-Process"
The first two steps are "physical isolation" and "process closure," while the third step is "data proof."
Our room design embeds "verifiable" DNA from the outset. From environmental monitoring point strategy, equipment surface sterilization verification interfaces, to critical room pressure differential logic and airflow visualization (smoke pattern test) design, all are aimed at meeting the most stringent requirements for aseptic process simulation verification (media filling).
Core Monitoring System:
Deploy high-density online environmental monitoring sensors to monitor in real-time the following areas: inside isolators, critical rooms, lyophilization channels, etc.:
- Particle count (≥0.5μm and ≥5μm)
- Airborne bacteria
- Settling bacteria
- Pressure differential
- Temperature and humidity
All data is automatically collected, tamper-proof, and bound with batch information in the Manufacturing Execution System (MES).
Establish Electronic Batch Environmental Records (EBER), enabling each batch of production to be associated with a complete environmental "health report," meeting FDA 21 CFR Part 11 requirements for data integrity and providing the ultimate electronic evidence for sterility assurance.
When auditors ask again: "Can you prove that every second of the production process for this batch of medicine was in a controlled environment?"
The answer is: Yes. The system records environmental data for every second, which can be retrieved and viewed.
V. Real Case Study: The Transformation of an East China Pharmaceutical Company
Dr. Wang's story has a continuation.
After experiencing one media fill positive result, they found Keyao. We designed and built a new fully enclosed isolator filling line and automated lyophilization integrated workshop for this innovative biopharmaceutical company in East China.
Data-Driven Results After Renovation:
1. Media Fill Test: From Positive to Zero Contamination
After adopting the isolator filling system, three consecutive Media Fill Test results were all "zero contamination," sterility assurance level was definitively proven, and market approval inspection was successfully passed.
Dr. Wang later told me: "When I saw the report showing zero contamination for three consecutive batches for the first time, my heart finally settled."
2. Lyophilization Automation: 30% Efficiency Improvement
Integrated automated lyophilization loading and unloading system enabled products after filling to enter the lyophilizer seamlessly and without personnel intervention under Grade B background protection, eliminating manual transfer risks and improving production efficiency by 30%.
Previously, two people working together could only complete loading and unloading for 2 batches per day; now, one person operating the AGV can easily complete 3 batches per day.
3. Data Traceability: Worry-Free Audits
Deployed a complete Environmental Monitoring System (EMS) and batch production process tracking system, capable of real-time retrospective tracing of environmental parameters (particles, airborne bacteria, pressure differentials, etc.) throughout the production lifecycle of any batch, receiving high praise in subsequent international client audits.
When the FDA inspector returned, we retrieved the full-cycle environmental data curve for that batch on-site. The inspector looked at it for a while and nodded: "This design is correct."
VI. Is This Solution Right for You?
If your situation matches the following characteristics, this method is likely suitable:
✅ Researching or producing high-value biologics such as monoclonal antibodies, recombinant proteins, peptides, and new vaccines
✅ Products use vial lyophilized powder injection form
✅ Currently in Phase III clinical sample production or commercial上市 filing phase
✅ Facing strict FDA, EMA, or NMPA inspections
✅ Have mandatory requirements for Sterility Assurance Level (SAL)
✅ Traditional open Grade A laminar flow sterile risks are difficult to control
Investment and Return Reference
| Project Scale | Suggested Investment Range | Expected Improvement | Payback Period |
|---|---|---|---|
| Single filling line (new) | 1.5-2.5 million | MFT first-time pass rate >80%, SAL improved | 6-8 months |
| Single filling line (renovation) | 0.8-1.5 million | Eliminate lyophilization transfer risk, data integrity compliant | 4-6 months |
| Complete lyophilization workshop (including lyophilizer channel) | 3-5 million | Full-process clean pathway closure, 30%+ efficiency improvement | 8-12 months |
Core Logic:
The loss from one sterile contamination causing batch scrapping could be hundreds of thousands or even millions. The time cost from one MFT failure causing filing delay could be months or even longer.
The root causes of these problems are often not operational errors, but outdated design philosophies.
VII. How to Get Started?
Keyao has specialized in cleanroom engineering for 13 years, is a council member of the Guangdong Cleanroom Industry Association, holds multiple invention patents and utility model patents, and has served over 100 clients.
If you are also facing sterile filling contamination risks or lyophilization process verification challenges, we have three steps to help you:
Step 1: Free Technical Assessment (3 days)
We will send engineers to your site to evaluate existing room design, identify sterility risk points, and issue a "Sterile Room Risk Diagnostic Report."
Step 2: Solution Design (1-2 weeks)
Based on assessment results and your budget, we provide 1-3 renovation plans, including:
- Basic Plan: Core area isolator renovation to solve the most critical filling risks
- Standard Plan: Isolator + lyophilization automation channel to eliminate lyophilization process risks
- Complete Plan: Full-process automation + intelligent monitoring system to achieve maximum SAL
Each plan includes: investment budget, implementation cycle, expected results, and ROI analysis.
Step 3: Project Implementation (4-12 weeks)
Keyao has EPC general contracting capabilities, providing integrated delivery from design, construction, and commissioning to certification support.
In the Pearl River Delta region, 2-hour response time, fastest 4 weeks to complete isolator system renovation, 8 weeks to complete complete workshop delivery.
Your Workshop Deserves Better Sterility Assurance
The essence of sterile pharmaceutical production is to achieve and continuously prove the absolute reliability of "zero microbial contamination" within complex human-machine-material-method-environment interactions.
This cannot be achieved simply by strengthening training or increasing inspections.
It requires shifting from a "personnel self-discipline" to an "engineering determinism" barrier protection philosophy.
We firmly believe: using engineering technology to reduce the contamination risk probability of the sterile core area to infinitely close to zero, creating a seamless production flow "from filling to capping under controlled protection," eliminating risk blind spots at process interfaces, and ensuring the continuity of sterile conditions.
This is our commitment to our clients and the core of our delivery of "worry-free workshops" that can withstand the most rigorous audits.
📞 Contact Us
If you are facing sterile filling room design, renovation, or certification challenges, please feel free to consult:
☎️ +86 139 2995 0401
(Monday to Friday 9:00-18:00)
💬 Follow the WeChat official account "Guangdong Keyao Purification" and reply "Sterile Room" to get a free technical assessment
🌐 Website: www.gdforyou.com
📊 Project Results Showcase

About Keyao Purification
Guangdong Keyao Purification Electromechanical Engineering Co., Ltd.
- Specialized in cleanroom engineering for 13 years
- 100+ successful cases
- 10+ technical patents
- Council member of Guangdong Cleanroom Industry Association
- "Contract-abiding and Trustworthy" Enterprise
Service Areas:
- Medical Cleanrooms: Surgery departments, ICU, NICU, DSA, Pharmacy Intravenous Admixture Services (PIVAS), Reproductive Centers, and over a dozen other special departments
- Industrial Cleanrooms: Biopharmaceuticals, health supplements, electronics, optics, medical devices, food and cosmetics workshops
- Laboratory Cleanrooms: Microbiology laboratories, PCR laboratories, animal facilities, physical and chemical laboratories, etc.
Our Clients:
Guangdong Second Provincial General Hospital, Dermatology Hospital of Southern Medical University, Foshan Traditional Chinese Medicine Hospital, Huarun Sanjiu Medicine, Yangtze River Pharmaceutical, By-Health, Darlie Group, and more.
The cases mentioned in this article are real projects with client information anonymized.